Methodological Strategies
in Microbiome Research and
their Explanatory Implications
Maureen A. O’Malley
University of Bordeaux
University of Sydney
Derek J. Skillings
University of Bordeaux
Early microbiome research found numerous associations between microbial
community patterns and host physiological states. These findings hinted at
community-level explanations. “Top-down” experiments, working with whole
社区, strengthened these explanatory expectations. 现在, “bottom-up”
mechanism-seeking approaches are dissecting communities to focus on specific
microbes carrying out particular biochemical activities (例如, choline metabo-
lism pathways, Clostridium difficile suppression). To understand the inter-
play between methodological and explanatory scales, we examine claims of
“dysbiosis,” when host illness is proposed as the consequence of a community
状态. Our analysis concludes with general observations about how methodol-
ogies relate to explanations, and the implications for microbiome research.
Introducing Microbiome Research
1.
Microbiome research is the analysis of the aggregated molecular compo-
nents of a defined microbial community (“microbiota”).1 Our examination
of this field will focus on how causality is assigned in microbiome analyses,
We are grateful for comments from discussion groups at Dalhousie University and the
University of Sydney, as well as from one of our anonymous referees. MAO is supported
by the French government’s “Investments for the Future” Programme, IdEx Bordeaux
(ANR-10-IDEX-03-02). DJS is funded by the European Research Council (ERC) 在下面
the European Union’s Horizon 2020 research and innovation programme—Grant agreement
no 637647—IDEM.
1. Although it was once common to distinguish the microbiota (the organisms) 从
the microbiome (their collective genome), current practice is often to refer to both organ-
isms and their molecular bases as microbiomes. See Prescott (2017) for a discussion of
the confused history of these terms. We will follow the older and narrower convention,
科学观点 2018, 卷. 26, 不. 2
© 2018 由麻省理工学院
土井:10.1162/POSC_a_00274
239
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
240
Methodological Strategies in Microbiome Research
and what methodological strategies are used to identify communities or
components of those communities as causal contributors to host states. Earlier
microbiome research was conducted at a broad scale via bioinformatic anal-
yses of environmental samples, then backed up by whole community transfer
实验. These “top-down” findings, in which the whole community is
treated as a unit of investigation, indicated that communities might act as
causal agents. 最近, as microbiome research has reconnected with
traditional experimental methods, communities have been decomposed in
the “bottom-up” search for traditional causal agents, such as single organisms,
or small groups of them, and their biochemical activities. These top-down
and bottom-up methodological strategies do not, 然而, map straight-
forwardly onto top-down or bottom-up explanations. This incongruity has
implications for the claims that can be made about the locus of causal action.
In the following discussion, we outline some developments in how
causality is investigated in microbiota-host research, and what the impli-
cations of various methodologies are for how these systems feature in
explanations. We first trace how microbiome research (especially that based
on human gut microbiota) has developed historically through association
学习, in which community patterns have been linked to broader system
outcomes such as the health and disease states of human hosts. We then
discuss recent research that returns the field’s focus to traditional targets of
microbiological experimentation: populations and specific biochemical
pathways. We illustrate this shift with brief accounts of new mechanistic
insights into choline metabolism and Clostridium difficile therapy. 我们的
analysis then turns to the explanatory conundrum of “dysbiosis,” in which
claims are made about host disorders as causal products of community-
scale changes in microbiota. We suggest that understanding how method-
ologies work at different scales can revise superficial impressions about the
explanatory and conceptual implications of microbiome research.
2. Historical Background to Microbiome Research
Microbiome research is a recent development in the life sciences. Its roots
lie in microbiology and molecular genetics, but its implications reach
beyond what is traditionally seen as microbiological subject matter. 在
its historically classic form, microbiology is a laboratory-based science
focused on pure cultures of microorganisms. Pure culturing methods be-
came so central to microbiology because of their experimental credentials
but quite a lot of the literature we cite does not. The broader use of the term sometimes
invokes the much older ecological meaning of “biome” to justify the organismal application
of microbiome). We will return to the potentially ecological interpretations of “micro-
biome” in Section 5.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
241
in establishing microbial causality via Koch’s postulates. The latter de-
scribe a sequence of isolating, culturing, and returning microbes to hosts.
They aim to demonstrate specific and consistent microorganismal effects,
especially those implicated in disease and food spoilage (Gradmann 2000;
Mendelsohn 2002; Ross and Woodward 2016). Biochemistry, as it rose
to prominence in microbiology in the early twentieth century, 更远
narrowed causal attributions by identifying the biochemical pathways by
which single lineages of microorganisms or small groups of them produce
effects (Singleton and Singleton 2016). Organism-specific activity has thus
been the traditional locus of causal assignment in microbiology.
At the same time that the laboratory-based pure culture approach
became established in the late nineteenth century, another set of practices
发达. It focused on microorganisms in natural environments (看
O’Malley 2014). This field also took a biochemical approach, and in the
middle of the twentieth century productively encountered molecular
genetic methods. In the 1980s, molecular efforts to understand micro-
organisms in their natural environments took a transformative turn. Norman
Pace and colleagues looked beyond the genes of isolated cells in laboratories.
They applied the maturing technology of DNA sequencing to in situ samples
of microbial communities (例如, Stahl et al. 1985; Olsen et al. 1986). Most of
these early environmental studies focused on genes for ribosomal RNA
(rRNA), which had already been established as useful markers for evolu-
tionary analyses (Woese and Fox 1977).
As evolutionary and ecological molecular approaches gathered momen-
tum in microbiology, sequencing technologies and bioinformatic tools
were also developing in both scope and speed. Entirely new pictures began
to emerge of previously unrecognized microbial diversity (例如, Mullins
等人. 1995). Culturing had for the time being reached its limits in reveal-
ing microbial diversity in many environments, often because the “pure”
growth of unknown microorganisms was obstructed by the complex inter-
dependencies in their communal lifestyles (Amman et al. 1995). 这
sequencing of a very large fragment of DNA in a marine sample, followed
by identification of the uncultured organismal group to which it belonged
(Stein et al. 1996), was confirmation that not only was this sequence-
based methodology feasible but also that it would produce novel findings.
This approach made an unexpected discovery of proteorhodopsin, a light-
reactive protein in marine bacteria, which was then confirmed experimen-
tally as functionally active (Béjà et al. 2000). The proteorhodopsin study
demonstrated very effectively that environmental sequencing of commu-
nity samples could lead to major discoveries, and that these could then
be interrogated further by classic scientific methods (see O’Malley 2008
for an overview).
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
242
Methodological Strategies in Microbiome Research
With ongoing improvements in sequencing technology and large data-
scale analysis, attention shifted from single genes (例如, rRNA) to whole
基因组. When these analyses targeted “environmental” genomes (IE。, 这
DNA extracted from samples collected from a range of natural environ-
评论), metagenomics was born. The first use of “metagenome” was in
a manifesto for soil metagenomics in 1998 (Handelsman et al. 1998,
p. R245). In that paper, molecular biologist Jo Handelsman and her
colleagues reflected on the untapped diversity of soils, which are a major
reservoir of medically and industrially important microbial compounds.
The new century saw metagenomics flourishing, as DNA from a very wide
range of environments and communities was sequenced and partly analysed
(Handelsman 2004).
Two quite different projects spurred on the field as they showed re-
searchers the potential of metagenomic tools. One was the metagenomic
analysis of the Sargasso Sea, a huge data-collection exercise that revealed
the extraordinary genetic diversity of microbial communities even in low-
nutrient environments (Venter et al. 2004). The other project was the analy-
sis of a still more restrictive environment: the acidic and anoxic runoff from
a mine (Tyson et al. 2004). Identification of all the organisms in this low-
diversity community was possible in the latter case; in the former, 还有
as assessments of population genetic structure, the function of genes (包括-
ing the light-reactive proteins discovered in earlier metagenomic samples)
could be tentatively assigned to uncultured organisms and inferences made
about their ecological roles.
As this new molecular field of microbial diversity studies grew, 注意力
increasingly turned to the human body as an ecosystem in its own right.
The potential of metagenomic tools was suggested for “the second human
genome”—the microorganisms untouched by the achievements of the
high-profile Human Genome Project (Relman and Falkow 2001, p. 206).
The gut metagenome in particular was recognized as an immense organ-like
source of genes and enzymatic activities that were mostly unknown, 和
likely to remain so, if laboratory-based culturing studies remained the only
option (例如, Eckburg et al. 2005; Zoetendal et al. 2004). Many gut microbes
can be cultured, but their full diversity (especially of anaerobes), plus their
互动, were and still are not understood in detail. To develop this
理解, international research consortia began to treat the human
body as constituting an ecological niche that with the gut microbiota forms
a complex multilevel system (Turnbaugh et al. 2007; O’Malley et al. 2014).
This ecological perspective began to influence an expanded interpretation of
the term microbiome, which was often taken now to emphasize “biome” and
its ecological connotations rather than “ome” and its more restrictive molec-
ular interpretation (see footnote 1 for terminological debates).
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
243
Although non-human ecosystems are at least as interesting and impor-
tant as human ones, there is no denying that the sheer amount of research
on the human microbiome, and particularly the gut microbiome, make it
an obvious focus for tracking how microbiome research has developed and
how causal inferences have been made in that development. The vast
majority of human microbiome literature focuses exclusively on bacteria,
which are the most abundant forms of life in the gut. This is done not only
for pragmatic reasons of reducing the complexities of analysis (IE。, 离开
out viruses and other unicellular organisms), but also because the greater
biochemical diversity in bacteria is thought to have more impact on the
human body. But as well as sequencing the gut community genomes
(or genetic markers in those genomes), then seeking patterns in that data,
human microbiome projects began to explore causal claims on the basis of
association studies. Causality was attributed to communities, 哪些是
initially discussed as if they were causally cohesive in the effects they
brought about in hosts.
3. Descriptions and Causes: From Pie-charts to Mouse Models
A great deal of early research on the human gut microbiota and its micro-
biome produced compositional “pie-charts” (as did microbiota studies of the
skin, mouth and other niches). The charts map the DNA sequence cate-
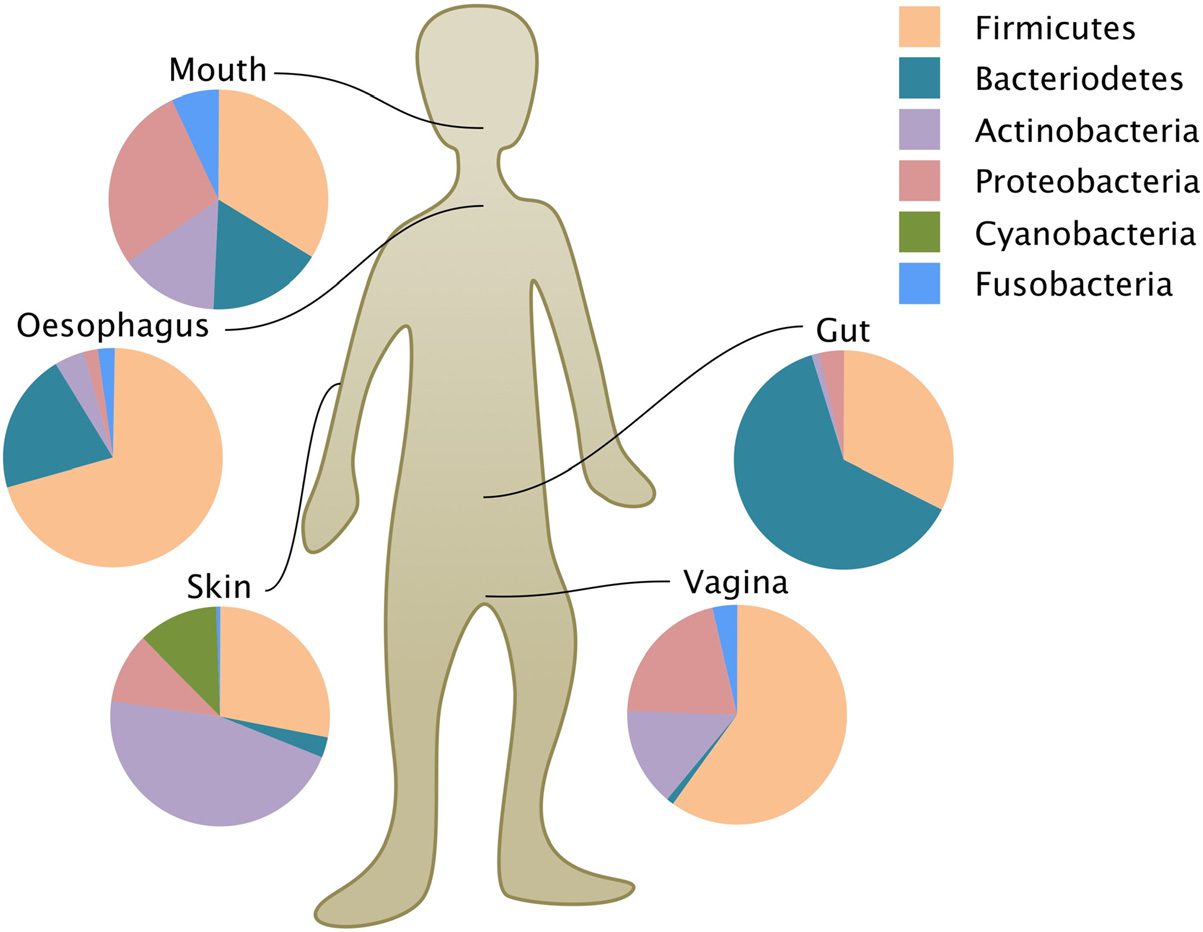
gorizations in the microbiome of interest (数字 1). These distributions of
sequence abundances are commonly presented at the phylum level. 尽管
organismal groups being identified in this categorization, they are rarely
characterized beyond the molecular markers that identify them. 虽然
there is extremely fine-grained microbial diversity at the species and strain
level in any single human body, it is much simpler to analyse molecular
patterns at the coarse phylum level. Phyla (sometimes called “divisions”),
are a very high level of taxonomic rank, just below kingdoms, 以上
orders, 类, 家庭, genera, and species (in descending order of the
taxonomic hierarchy). There are anywhere between 30 and a thousand bac-
terial phyla recognized by different methods and criteria.2 The great majority
of healthy human guts so far sampled are dominated by only two phyla:
Firmicutes and Bacteroidetes (数字 1). Together they comprise about
90% of human microbial gut diversity.
2. The phylum rank is not recognized by official classification nomenclature (Parte
2013), and it is not clear what the boundaries for a prokaryote phylum would be, 或者
how many bacterial phyla there might be, especially with burgeoning environmental dis-
coveries of putative bacterial phyla (例如, Yarza et al. 2014; Brown et al. 2015). Recent
microbiome classification pushes toward lower taxonomic levels (例如, Johnson et al.
2017; Duvallet et al. 2017), but broad-brush pictures are still the norm.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
244
Methodological Strategies in Microbiome Research
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
数字 1. A variety of human microbiomes (the molecular compositions of
particular communities) represented proportionally at the phylum level by
pie-charts (based on Spor et al. 2011; adapted by Michel Durinx (https://centimedia.
组织/) and used with permission from Nature/Springer/Palgrave, 自然评论
Microbiology, copyright 2011).
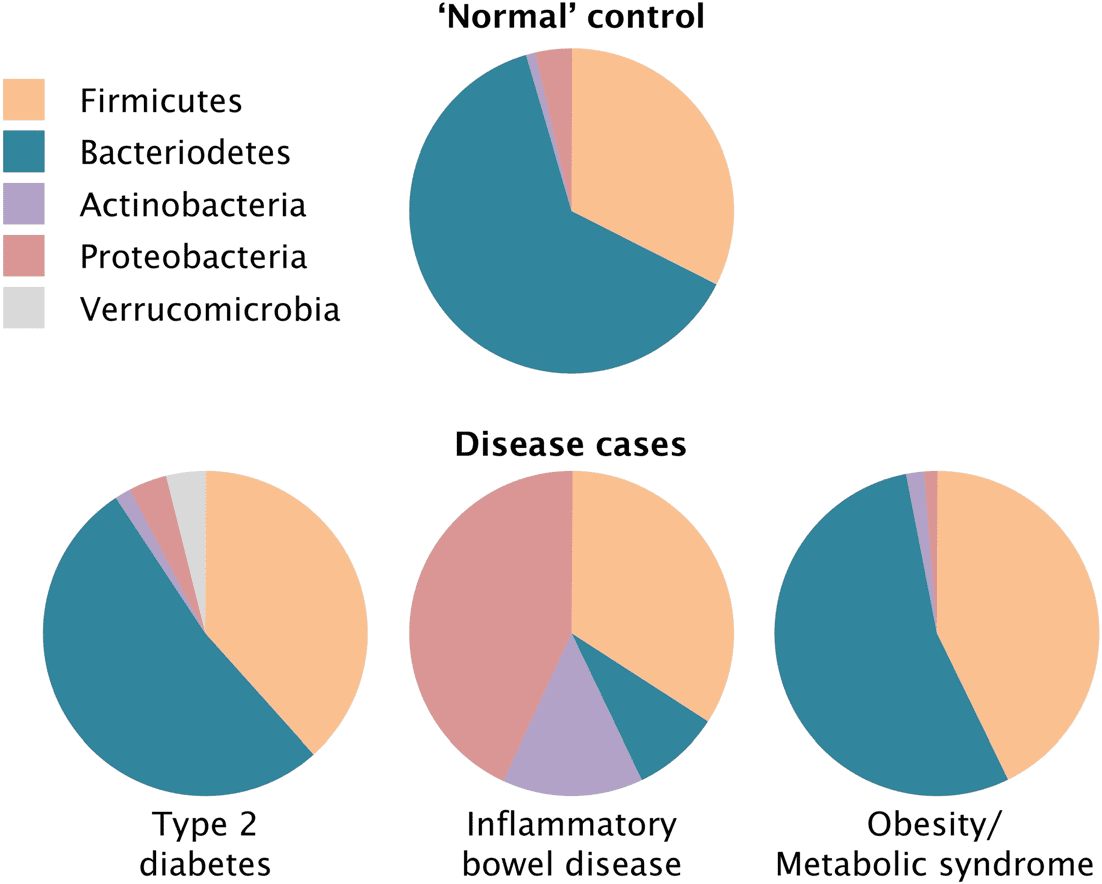
Once the composition of a microbiome—such as that of the human gut—
has been roughly described and categorized, these patterns can be “linked”
to or associated with particular health and disease states. 比较
with microbiomes in humans lacking the disease indicates visually how
phyla patterns are different in diseased hosts (数字 2). Although these
comparisons are done for numerous body niches, it is the distal gut—the
large intestinal colon—that has been the focus of the most work (Marchesi
2011). This trend has been driven partly by the ease of access to gut micro-
biota diversity, which is commonly via faecal samples. Although there are
questions about the adequacy of this representation,3 by and large this
representativeness has been and still is taken for granted.
For some important diagnostic and predictive purposes, phyla pro-
portions may potentially tell researchers about disease states. Numerous
3. The composition of microbiomes sampled via faeces versus intestinal biopsies are
often not concordant (Momozawa et al. 2011). There are indications that samples taken
directly from the gut will more accurately predict disease states (Gevers et al. 2014).
科学观点
245
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
数字 2. A typical representation of the human gut microbiome and associations
between broad compositional patterns and disease (based on Spor et al. 2011; adapted
by Michel Durinx (https://centimedia.org/) and used with permission from Nature/
Springer/Palgrave, 自然评论微生物学, copyright 2011). Sometimes only a
few samples underpin such patterns, which is statistically very low-powered given
how much inter-individual variation exists between each human’s microbiome.
Ideally such associations should be interpreted as patterns worth exploring, 和
not as statistically robust correlations, but sometimes stronger interpretations
被制作.
学习, including some formative microbiome research, have shown that
an elevated proportion of Firmicutes is associated with host obesity and the
metabolic syndrome accompanying it (例如, Ley et al. 2006; Turnbaugh
等人. 2006, 2008). Phylum-level patterns associated with conditions such
as human obesity have been described as “highly conserved bacterial traits”
that affect host phenotype (Ley et al. 2006, p. 1023). Although obesity
investigations are emblematic of human microbiome association studies,
a wide range of other diseases, including brain and behavioural disorders
(Bercik et al. 2011; Hsiao et al. 2013), have now been associated with
coarse changes in microbiota diversity. Many of these association studies
不要, 然而, go on to examine any causal implications very thoroughly
(IE。, whether changes in composition are causes or effects of disease states).
Knowledge of associations can in some cases be backed up by experi-
mental regimes, in which entire microbiota are transferred into animal
246
Methodological Strategies in Microbiome Research
hosts, mostly mouse models. Led by early efforts in Jeffrey Gordon’s lab,
microbiome researchers began to manipulate microbiota experimentally.
This work involves “germ free” laboratory mice. They are born by caesarean
section and raised in sterile environments (including irradiated food) so that
they are not colonized by microbiota, unlike conventionally born and reared
老鼠. Microbiome researchers transplant microbiota samples—commonly via
faeces from conventional mice with specific phenotypic properties, 乃至,
出奇, from humans4—into these germ-free model mammals, 和
then track physiological changes in order to scrutinize cause-effect relation-
船舶 (Turnbaugh et al. 2006, 2009; Ridaura et al. 2013). Researchers have
frequently found that transplanting phyla-differentiated microbiota into
mice can make a major physiological difference to the host. 换句话说,
transmission of phenotype at the organismal (老鼠) level can occur via
transmission of the microbiota. In these cases, “the microbiome is considered
causal” (Goodrich et al. 2014, p. 250).
Although there are additional and possibly confounding causal factors
that must be taken into consideration (especially diet, and medications such
as antibiotics), research using mouse models and entire microbiota trans-
plants became the gold standard for causal attributions in microbiome stud-
是的. 再次, obesity has been a particularly successful example of
phenotypic transfer by microbiota transplant, confirming the causality
inferred from bioinformatic associations (例如, Turnbaugh et al. 2009).
Firmicutes-Bacteroidetes proportions, or other broad changes in community
diversity, continued to be implicated as significant causal contributors to
obesity and other diseases.
But as microbiome research has developed further, phylum-level propor-
tionality or decreased diversity turns out to offer less insight into human (和
老鼠) obesity and other conditions than originally anticipated (Duvallet
等人. 2017; Finucane et al. 2014; Sze and Schloss 2016; Walters et al.
2014).5 Both increases and decreases of key phyla may be associated with
obesity. 更糟, experimental replications have contradicted previously
postulated effects of microbiota changes (Fleissner et al. 2010; Harley and
Karp 2012; Schwiertz et al. 2010). One response is to see if consistent obser-
vations can be achieved by increasing sample size (例如, Beaumont et al. 2016;
Falony et al. 2016). Another is to look within the community for smaller
scale causal agents.
4. This is surprising because of the phylogenetic specificity of many microbiota-host
关系 (Chung et al. 2012; 然而, 比照. Seedorf et al. 2014, which shows some un-
expected flexibility).
5. Small sample sizes and their statistically underpowered findings play a role here,
as noted in section two.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
247
4. From Top-down to Bottom-up Microbiome Analyses
Establishing fine-grained causal relationships has become for many micro-
biome researchers the driving aim of the field (Fischbach and Segre 2016;
Gilbert et al. 2016; Surana and Kasper 2017). This wave of research
focuses on pinpointing, 例如, “the specific bacteria exclusively
associated with obesity” and other health-disturbing syndromes (Harley
and Karp 2012; 张等人. 2009, p. 2365; 赵 2013). Some researchers
describe efforts to carry out finer grained experimental-mechanistic work as
“bottom-up” microbiome research, in order to distinguish it from the “top-
down” methodologies discussed in Section 3. The latter are driven by com-
putational analyses and pattern-seeking approaches, then supplemented
by whole-microbiota transplants to mice (例如, Huttenhower et al. 2014,
Macpherson et al. 2015, Moya and Ferrer 2016).6 然而, many bottom-up
approaches are based on or include top-down approaches that treat the
community as a whole before doing traditional cause-establishing research.
New microbiome research on choline metabolism7 and Clostridium difficile
therapy show how these emerging decompositional approaches work.
4.1. Choline Metabolism
Choline is an essential human dietary nutrient found primarily in red meat
and egg yolks. Gut microbes convert some of it anaerobically to trimethyl-
amine, which is then oxidized in the liver. Trimethylamine in this oxidized
form is associated with cardiovascular problems in humans and mice (唐
and Hazen 2014).8 An important step in unravelling this causal chain
showed that mice fed with choline tend to develop atherosclerosis (难的-
ening of the arteries), and more precisely, that suppressing all gut microbes
in mice inhibits this disease (Wang et al. 2011). But rather than attribute
causal agency to the microbiota generally or to certain proportions of
phyla, the next research step zoomed in on a gene cluster of a sulphate-
reducing gut bacterium. This organism could be cultured and manipu-
lated genetically (Craciun and Balskus 2012). Researchers then went on
to characterize the exact mechanism of how this bacterium metabolizes
choline. They noted that their discovery “shows the potential of combining
analysis of a biochemical mechanism with bioinformatic analysis” (Craciun
6. Not all these citations refer to exactly the same combinations of approach, 但他们的
distinctions are broadly consistent. We include the mouse microbiota transplants in the
top-down category, because these experiments are transferring the whole community and
looking for host-wide effects.
7. Thanks to Andrew Roger (Dalhousie) for suggesting this example.
8. A few studies show no connection between choline intake and atherosclerosis, 但
they are in the minority.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
248
Methodological Strategies in Microbiome Research
and Balskus 2012, p. 21310), so that specific causal agents can be localized
in broader microbiome patterns. This combination of top-down and
bottom-up methodologies also worked for studies that tracked the conver-
sion of another red meat substance, L-carnitine, to trimethylamine via gut
microbes (see Falony et al. 2015 for an overview).
What appears to be happening in the choline research area is a tradi-
tional decompositional analysis, in which microbiota systems are broken
down to their parts, and methods applied to identify the details and order-
ing of causal effects (for a list of other examples, see Fischbach and Segre
2016). This classic approach aims to fill in the mechanistic links in a causal
链. Other interactions exist, 为了确定, but they are given background
地位. 例如, when the choline metabolism process is tracked by
experiment and biochemical inference to identify a pathway in an organ-
主义, it does not mean that the production of trimethylamine is necessarily
confined to a single organism (see Zhu et al. 2016) but that any additional
choline metabolizers need to be studied separately.
Other areas of research, particularly concerned with how the human
immune system develops in concert with microbiota composition and func-
的, also show considerable promise for filling in detailed mechanistic steps
between microbiota patterns and human disease (Round and Mazmanian
2009; Hooper et al. 2012). Both biochemistry and immunology are exper-
imentally focused sciences, and they appear to be drawing on large-scale
bioinformatic analyses primarily in order to stimulate hypotheses and their
experimental investigation.9 But not all microbiota research works in this
方式; some of it shows consistent effects from community-level intervention.
尽管如此, even in these cases, there is explanatory pressure to “down-
size” causality by pinning causal interactions to the organismal level.
4.2. Clostridium Difficile Therapy
One of the most dramatic success stories in microbiome research comes
from the use of faecal microbiota transplants (FMTs) as treatments for
Clostridium difficile infections in human intestines. This organism, 哪个
is often carried asymptomatically and only becomes pathogenic after anti-
biotic treatments (especially post-surgery), can cause long-term illness,
pain and eventually death to infected humans. C. difficile forms spores,
which enable transmission are also resistance to antibiotic therapies against
有机体. More than four decades of studies of various degrees of rigour
and duration have shown that microbiota transplants from healthy humans
9. This combination of top-down and bottom-up methods is why the proteorhodopsin
story was such a powerful motivator for metagenomic research (参见章节 2).
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
249
can suppress C. difficile activities and effects (Kassam et al. 2013; van Nood
等人. 2013). The relative proportion of Firmicutes and Bacteroidetes is re-
portedly “restored or ‘rebalanced” in the receiver’s gut by microbiota
移植, and the various symptoms of C. difficile infection disappear or
ameliorate as this general state of “dysbiosis”—an unhealthy composition of
the community—is remedied (Borody and Khoruts 2012).
然而, even though this intervention is done at a community scale
(IE。, the entire microbiota, or at least as many as survive passage from one
human body to another via faecal transplant), for some scientists this strat-
egy is seen as a stop-gap measure rather than an intervention that suggests
causal efficacy at the community level. There has already been some success
in decomposing the “anti-dysbiotic” FMT microbiota, experimentally and
mathematically by “rational design.” This work targets the precise causal
agents that ameliorate C. difficile infections (例如, Buffie et al. 2015; Lawley
等人. 2012, Almeida et al. 2016). In a particularly powerful combination
of methods—biochemical experiments, bioinformatic analysis, and math-
ematical modelling—one of these studies identified the secondary bile acid
synthesis carried out by certain organisms (other Clostridium species, 埃斯佩-
cially C. scindens) as the specific biochemical mechanism by which C. difficile
growth is suppressed (Buffie et al. 2015).
The identification of finer grained causality in bottom-up studies is
done not just for the sake of basic scientific explanation, but also to develop
therapeutic treatments that exploit these discoveries of causal pathways.
Despite the successes of community-level FMT interventions on C. difficile
infections, drug development efforts are based on the belief that causality
should be attributable to identifiable lineages, and to isolatable pathways
in those lineages. These “bottom-up” accounts of causal agency in micro-
biota research thus attempt to follow quite traditional scientific practices
in microbiology, even when initial efforts were able to make loose causal
ascriptions at the higher community level.
5. Dysbiosis and Homeostasis in Microbiome Explanations
Constructing causal explanations is a key motivating activity for scientists.
Descriptions of these causes are part of the standard mechanistic explanatory
apparatus that is taken for granted as the basis of good science.10 Decompo-
sitional analyses are usually what enable mechanistic causal attributions
(Bechtel and Richardson 1993). Retaining a causal focus on communities
might thus seem unlikely to continue as the field develops. 然而, 这
10. The same is not true of much medical research, especially “evidence-based medicine,”
where evidence for mechanisms is often very low down in the evidence hierarchy (安徒生
2012; Howick 2011; La Caze 2011).
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
250
Methodological Strategies in Microbiome Research
microbiome story is not necessarily one of a broad-brush view being right-
fully replaced by a more detailed and complete one as the field develops.
Microbiome researchers continue to hint at or even argue explicitly for
community-level causation that might resist such decomposition.
One of the most obvious ways this occurs is when microbiome researchers
attribute “dysbiosis” to the community level, by reasoning that “some diseases
might result from dysbiosis rather than the presence of a single disease-causing
microbe” (Clemente et al. 2012, p. 1263). Such claims are justified by beliefs
that “it is likely that no one single organism will work most effectively, 但
rather a complex assortment of organisms will provide the maximum benefit”
to the host (Petersen and Round 2014, p. 1030). Dysbiosis in microbiome re-
search is defined very loosely, and refers broadly to a “compositional or func-
tional shift within host-associated microbial communities that has the
potential to facilitate growth of pathogens and/or [这] onset [的] diseases”
(Arnold et al. 2016, p. 889; see Hooks and O’Malley 2017 for a critique).
Changes to phylum ratios or general decreases in diversity commonly
serve as markers of dysbiotic alterations (Lewis et al. 2015). These changes
are detected post-hoc, when hosts known to be diseased exhibit “change
to the composition of resident commensal communities relative to the
community found in healthy individuals” (Petersen and Round 2014,
p. 1024). 然而, this ideal or normal state is usually unspecified, or sim-
ply described as having more diversity.11 Moreover, it is usually impossible
to discern whether inferred dysbioses occur prior to the disease they are pu-
tatively causing or afterward, as a consequence of the disease (Bäckhed et al.
2012). There may also be common causes, such as diet or inflammation,
that produce both the altered microbiota composition and the disease.
When dysbiosis is proposed as a system-level explanation of host phys-
iology, it is often implied that there is a “balanced” state to which the
whole microbiota normally contributes. This balance is thought to arise
from the evolved normalness or optimality of the sum of interactions be-
tween human bodies and their microbial commensals (例如, Belkaid and
Hand 2014; Fuentes et al. 2014, Reid et al. 2011). It is common in
microbiota research to refer to this supposedly balanced state as one of
“homeostasis,” although sometimes the terms “eubiosis” and “normo-
biosis” are used (例如, LePage et al. 2013; Schulberg and De Cruz 2016).
Just as for claims about dysbiosis, homeostasis is asserted very casually
and loosely, as something “normal” and physiologically desirable (例如, 里德
等人. 2011). Quantified theoretical claims, such as might accompany tra-
ditional cybernetic views about homeostasis, are not attempted.
11. 看, 然而, Gevers et al. (2014), for an example of a more precise “dysbiosis index”
correlated with a particular disease state.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
251
What do bottom-up methodological developments in microbiome
research mean for dysbiosis and homeostasis and their implicit and
explicit claims about community-level causation? And more generally,
why might changes in community-scale composition, especially different
proportions of phyla, even be thought of as plausible causal agents in the
first place? One answer comes from general perspectives on systems and
explanations at the system level. It is well known that system states can
produce effects, and that such relationships may not be revealed by sim-
ple experimentation that searches for and establishes single mechanisms
and linear causal chains (see Green et al. 2017).
A more substantive reason for continuing to focus on the community
as a causal locus is the extraordinary functional redundancy in commu-
nities such as those of the human gut. Knock out one species or strain,
and another will commonly supply the same products. Robust function
supplied by networks of genes and metabolic pathways may be the rel-
evant explanatory locus rather than the more transient individual tax-
onomic units that bear and share such genes (Doolittle and Booth
2016; Louca et al. 2016; Taxis et al. 2015). These networks will often
not be localized in single populations of organisms. Functional accounts
of how communities robustly bring about health or disease are now
seen as a way forward for community-based explanations of host state
(Knights et al. 2013; 利维等人. 2017; Moya and Ferrer 2016). Never-
theless, to understand these functional contributions, decompositional
analysis of the community still has to be done to show how members
work together to create functions that are distributed across different
lineages of organisms. Even when clusters of organisms from different
lineages are known to be causal players, knowledge of the individual
genes and pathways that make up that cluster is required by the cur-
rent methodological configuration and explanatory expectations of the
领域.
例如, network models based on high-throughput data (哪个
microbiome research has in abundance) are an important means of
making system-level explanations (Borenstein 2012; Faust and Raes 2012;
Greenblum et al. 2013; Layeghifard et al. 2017). 然而, revealing the
organizational structures of microbiome networks continues to be done on
the basis of associations (例如, Greenblum et al. 2012), and many network
explanations still require the identification of localized mechanisms (例如,
Noecker et al. 2016). A different strategy is to analyse community-scale
correlations with host states more rigorously in order to evaluate whether
causal claims can be justified (Cho and Blaser 2012; Gilbert et al. 2016).
Larger samples and more detailed analysis of the conditions that affect
microbiota composition will also help reframe and refine community
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
252
Methodological Strategies in Microbiome Research
association approaches (Falony et al. 2016), and thus potentially enable
system-level explanations of microbiome effects.
Many microbiome researchers also expect that community-scale ecolog-
ical models will eventually enable predictions and explanations of micro-
biota states and their effects (Costello et al. 2012; Coyte et al. 2015;
Marino et al. 2014). In plant and animal ecology, models of community
stability have been important for explanations of ecosystem dynamics
that range from collapse to flourishing (see Justus 2008; McCann 2000).
同样地, in microbiome research, the stability or instability of whole-
community composition is thought to be an explanatory candidate for
some disease states in hosts (例如, Jeffery et al. 2016). Community genetics,
usually applied to plant communities (Whitham et al. 2006),12 may also pro-
vide explanatory resources for microbiome explanations at the community
scale by showing how genetic variation in populations contributes to com-
munity structure and ecosystem behaviour (Skillings 2016).
An illustration of a rudimentary ecological explanation of microbiota
and host builds on the success of FMTs in “curing” C. difficile pathologies.
The influx of donor microbes is hypothesized to affect the niche structure
of the gut due to the transplanted microorganisms taking over niches that
C. difficile had usurped. This takeover also explains the diversity depletion
of the pre-transfer microbiota, which is strongly associated with C. difficile
感染 (Khoruts et al. 2010; Lawley and Walker 2012). Explanations like this,
while still sketches, could be fleshed out and provide genuinely community-
level ecological explanations. This fleshing-out, 然而, would require the
initial decomposition of the community to understand the functional roles,
niches, and interactions that led to the success of the transplant.
Although we think it likely that the future of microbiome explanation
will be ecological (and would thus emphasize the “biome” interpretation of
microbiomes—see Section 1), a great deal of methodological development
and detailed research is required before community-level hypotheses about
稳定, robustness and “dysbiosis” are established on a stronger evidential
and inferential knowledge base. For the time being, such hypotheses pro-
vide at best indications of scenarios that need further attention and might
eventually be filled in mechanistically. A key issue here is whether com-
munities treated as methodological targets will map onto the explanatory
targets of microbiome research.
Implications of Microbiome Research Methods for Explanation
6.
Earlier, we described how microbiology’s pure-culture approaches had to
isolate organisms from their complex communities in order to home in on
12. This form of community genetics is not the medical or public health version.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
253
桌子 1. Top-down and bottom-up interpretations. Although it looks as
if the entries in each column should work together, we suggest that
microbiome research may currently be restricted to explanations on the
bottom-up side, even when top-down methodologies are employed.
Current methodological and explanatory achievements are shaded.
Top-down
Bottom-up
Methodological target
Community patterns
Explanatory level
System states as
mechanisms
Individual organisms
and pathways
Single components
and mechanisms
Explanatory strategy
Non-decompositional
Decompositional
central relationships of cause and effect. 现在, microbiome research is in
search of explanations generated on the basis of findings from community-
level methods. It is not clear that current methodologies are necessarily
going to achieve explanations at the community scale (桌子 1). 当前的
restrictions on transforming community-level findings to explanations at the
same scale do not mean top-down attributions are irrelevant or redundant:
至少, top-down associations and experiments play a central role
in identifying broad and potentially causal relationships. But it is more tra-
ditional methods of decomposing those entities and isolating very specific
effects that seem to be picking up the epistemic baton for the microbiome
research community.
Early microbiome research, driven by bioinformatic analyses and top-down
实验, necessarily worked with and drew attention to community-
scale patterns (Huss 2014; Manor et al. 2014). But now, as more traditional
methods of experimentation are brought to bear on microbiota, the causal
focus often becomes a smaller system: an organism or population of organ-
isms that possesses particular biochemical pathways. For better or worse,
scientific confidence about causal claims tends to be built on experimental
manipulation of small-scale systems and their components (Craver 2006).13
Claims about dysbiosis might at first glance be thought to function as
explanatory place-holders for more substantive accounts of how com-
munity proportions or diversity generally affect host health. Particularly
when causality is localized to specific microbe-host interactions, 如
13. In biomedical research, where microbiome analysis has achieved considerable infil-
翻译, confidence comes from randomized controlled trials. Such trials are not anticipated
to play a large role in microbiome research’s immediate future, except for simple treatment
regimens involving probiotics and microbiota transplants.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
254
Methodological Strategies in Microbiome Research
the choline and C. difficile cases, suggestions of dysbiosis as the loss of
homeostasis or natural balance appear to be too loose and global to do
much genuine explanatory work. Relationships between microorganisms
and their human hosts might in some cases at least be explained by specific
interactions between individual organisms and pathways, 而不是
broad assertions about dysbiosis and homeostasis. This does not mean that
claims about community-level states such as dysbiosis are made false, 但
that the pressure is mounting for them to be evaluated more rigorously and
filled in mechanistically (例如, Hooks and O’Malley 2017; Olesen and Alm
2016).
然而, although mechanism-oriented research can indeed provide
causal explanations of particular phenomena, it is clear there are many in-
stances when causal complexity overwhelms straightforward mechanistic
账户 (安徒生 2012; Green et al. 2017). Even though microbial com-
munities may eventually be decomposed causally, explanatory payoff is
still expected at the larger scale, on the grounds that network or large-
scale ecosystem properties may achieve host effects. 然而, achieving
community-level explanations of host health and disease continues to be
dependent on knowledge about small-scale causal agents. Assigning cau-
sality in such situations and pursuing broad system-level mechanisms (例如,
putative dysbiosis states) still await the development and application of
appropriate methodologies and explanatory strategies in microbiome re-
搜索. 迄今为止, the maturation of microbiome research highlights how expla-
nations are not necessarily generated at the same scale as the methodological
starting point.
Implications for Conceptualizing Microbiota
7.
One important consequence of understanding the relationship between
methodological and explanatory strategies in microbiome research has to
do with the field’s implications for biological and evolutionary individ-
uality (this is a topic commentators from outside the field have found par-
ticularly interesting, although many of them have paid limited attention
to microbiome research practices). If microbial communities bring about
host effects primarily as a whole, then these systems might be understood
as closely integrated, functionally intertwined, mutually dependent groups of
有机体. 经常, claims that microbial communities form cohesive causal
entities flow into assumptions of evolutionary unity; such assumptions
are now seriously criticized (例如, Douglas and Werren 2016).14 然而,
14. We will not linger over these debates here, but do suggest they need viewing from
an explanatory angle.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
255
even when it is only physiological unity being considered, the degree of
cohesiveness between members in any community still needs to be worked
out with regard to specific causal interactions. Many important negative
and positive causal interactions will almost inevitably localize to populations
within those communities, and vary according to how those groups interact
differentially with other populations in the host environment (例如, Estrela
等人. 2016).
The strength of any interactions between populations is also dependent
on environmental variables such as diet, dispersal ability, 地理, 和
host physiology (Falony et al. 2016). These factors are unlikely to operate
on different populations in exactly the same way, nor on the relations be-
tween them. There will be different degrees of cohesiveness between com-
ponents (人口) of the microbiota. 因此, for many explanatory
目的, such as the investigation of specific health or illness states, 这
focus will not be the community as a whole. Each case will require careful
analysis of specific causal interactions, and assessments of whether micro-
biota activities are widespread or localized, negative or positive. There may
be some historical parallels here with plant community research of the
early part of the twentieth century. Notions of a highly organized commu-
本质 (a physiological unit) were eventually eclipsed by views that such en-
tities are more helpfully understood as structured assemblies of individual
organisms with specific causal inputs (Odenbaugh 2007). By understanding
these individual activities, insight into community dynamics is gained.
One factor that may have contributed to assumptions about the phys-
iological unity of microbiota is that early human microbiome analyses took
for granted the mutual benefits of human-microbiota relationships (例如,
Bäckhed et al. 2005; Hooper and Gordon 2001). Evolutionary accounts
have been given of these assumed mutual benefits. 例如,
The shared evolutionary fate of humans and their symbiotic
bacteria has selected for mutualistic interactions that are essential
for human health, and ecological or genetic changes that uncouple
this shared fate can result in disease. (Dethlefsen et al. 2007,
p. 811).
The underlying rationale is that because humans without microbes do
not exist, evolution must surely have worked out ways for humans to get
along positively, and even optimally, with their passengers. There should,
所以, be causally tight and ultimately beneficial relationships that
unify host and microbiota. This line of thinking informs views of dysbiosis:
perturb the evolved mutualistic relationship, and there will be bad con-
sequences for the whole system of host and microbiota (例如, Schwabe
and Jobin 2013; Wu and Lewis 2013).
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
256
Methodological Strategies in Microbiome Research
But as microbiota relationships are analysed more closely both theoret-
ically and experimentally, it is clear that ongoing relationships between
host and microbiota are very likely to be antagonistic, competitive and
exploitative rather than cooperative (Coyte et al. 2015). The balance-sheet
of cooperative and competitive relationships will vary over time and cir-
cumstance. Calibrating these relationships requires knowledge about
individual lineages, their functions, and how they interact (例如, Rakoff-
Nahoum et al. 2016). This ecological focus then informs evolutionary
accounts of the community. Even long-term evolutionary associations
between microbes and human hosts (plus other mammals) are limited
to a few microbial lineages (Grouissin et al. 2017), thus limiting what can
be said about the microbiota as a whole causal unit with selected effects on
the host.
In Summary
8.
Focusing on the interaction between methodology and explanation in
microbiome research may thus deflate expectations about groups of diverse
有机体 (such as hosts and their microbial communities) as unified bio-
logical or evolutionary entities. Identifying the actual locus of explanation
is central to understanding not just what microbiota are but what they do.
As we have argued, there are many ways in which explanation may be
pitched at the system level. Genuinely ecological approaches to explana-
tion can make sense of phenomena produced by communities through a
range of interactions, many of which have yet to be understood at the
relevant causal level. But for now, disentangling explanatory expecta-
tions from methodological achievements can help to understand what
the field of microbiome research has accomplished and where it might
potentially develop.
参考
Almeida, Rowena, Teklu Gerbaba, and Elaine O. Petrof. 2016. “Recurrent
Clostridium difficile Infection and the Microbiome.” Journal of Gastro-
enterology 51: 1–10.
Amman, Rudolf I., Wolfgang Ludwig, and Karl-Heinz Schleifer. 1995.
“Phylogenetic Identification and In Situ Detection of Individual Micro-
bial Cells Without Cultivation.” Microbiology Reviews 59: 143–169.
安徒生, Holly. 2012. “Mechanisms: What Are They Evidence for in
Evidence-Based Medicine?” Journal of Evaluation in Clinical Practice 18:
992–999.
Arnold, Jason W., Jeffrey Roach, 和M. Andrea Azcarate-Peril. 2016.
“Emerging Technologies for Gut Microbiome Research.” Trends in
Microbiology 24: 887–901.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
257
Bäckhed, Frederik, Ruth E. Ley, Justin L. Sonnenburg, Daniel A. 彼得森,
and Jeffrey I. Gordon. 2005. “Host-Bacterial Mutualism in the Human
Intestine.” Science 307: 1915–1920.
Bäckhed, Frederik, Claire M. 弗雷泽, Yehuda Ringle, Mary Ellen Sanders,
右. Balfour Sartor, 等人. 2012. “Defining a Healthy Human Gut Micro-
biome: Current Concepts, Future Directions, and Clinical Applications.”
Cell Host & Microbe 12: 611–622.
Beaumont, Michelle, Julia K. Goodrich, Matthew A. Jackson, Idil
然而, Emily R. Davenport, 等人. 2016. “Heritable Components of the
Human Fecal Microbiome are Associated with Visceral Fat.” Genome
生物学 17: 189.
Bechtel, 威廉, and Robert C. 理查森. 1993. Discovering Complexity:
Decomposition and Localization as Strategies in Scientific Research. 普林斯顿大学
新泽西州: 普林斯顿大学出版社.
Béjà, Oded, L. Aravind, Eugene V. Koonin, Marcelino T. Suzuki, 安德鲁
Hadd, 等人. 2000. “Bacterial Rhodopsin: Evidence for a New Type of
Phototrophy in the Sea.” Science 289: 1902–1905.
Belkaid, Yasmin, and Timothy W. Hand. 2014. “Role of the Microbiota in
Immunity and Inflammation.” Cell 157: 121–141.
Bercik, Premysl, Emmanuel Denou, Josh Collins, Wendy Jackson, Jun Lu,
等人. 2011. “The Intestinal Microbiota Affect Central Levels of Brain-
Derived Neurotropic Factor and Behavior in Mice.” Gastroenterology 141:
599–609.e3.
Borenstein, Elhanan. 2012. “Computational Systems Biology and In Silico
Modeling of the Human Microbiome.” Briefings in Bioinformatics 13:
769–780.
Borody, Thomas J., and Alexander Khoruts. 2012. “Fecal Microbiota
Transplantation and Emerging Applications.” Nature Reviews Gastro-
enterology 9: 88–96.
棕色的, Christopher T., Laura A. Hug, Brian C. 托马斯, Itai Sharon,
Cindy J. Castelle, 等人. 2015. “Unusual Biology Across a Group
Comprising More Than 15% of Domain Bacteria.” Nature 523: 208–211.
Buffie, Charlie G., Vanni Bucci, Richard R. 斯坦因, Peter T. McKenney,
Lilian Ling, 等人. 2015. “Precision Microbiome Reconstitution Restores
Bile Acid Mediated Resistance to Clostridium difficile.” Nature 205:
205–208.
给, Ilseung, and Martin J. Blaser. 2012. “The Human Microbiome:
At the Interface of Health and Disease.” Nature Reviews Genetics 13:
260–270.
钟, Hachung, Sünje J. Pamp, 乔纳森·A. 爬坡道, Neeraj K. Surana,
Sanna M. Edelman, 等人. 2012. “Gut Immune Maturation Depends
on Colonization with a Host-Specific Microbiota.” Cell 149: 1578–1593.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
258
Methodological Strategies in Microbiome Research
Clemente, Jose C., Luke K. Ursell, Laura Wegener Parfrey, and Rob
骑士. 2012. “The Impact of the Gut Microbiota on Human Health:
An Integrative View.” Cell 148: 1258–1270.
科斯特洛, Elizabeth K., Keaton Stagaman, Les Dethlefsen, Brendan J. 中号.
Bohannan, and David A. Relman. 2012. “The Application of Ecological
Theory toward an Understanding of the Human Microbiome.” Science
336: 1255–1262.
Coyte, Katherine Z., Jonas Schluter, and Kevin R. 促进. 2015. “这
Ecology of the Microbiome: 网络, Competition, and Stability.”
科学 350: 663–666.
Craciun, Smaranda, and Emily P. Balskus. 2012. “Microbial Conversion
of Choline to Trimethylamine Requires a Glycyl Radical Enzyme.”
Proceedings of the National Academy of Sciences U.S.A. 109: 21307–21312.
Craver, Carl F. 2006. “When Mechanistic Models Explain.” Synthese 153:
355–376.
Dethlefsen, Les, Margaret McFall-Ngai, and David A. Relman. 2007. “An
Ecological and Evolutionary Perspective on Human–Microbe Mutualism
and Disease.” Nature 449: 811–818.
Doolittle, 瓦. Ford, and Austin Booth. 2016. “It’s the Song, Not the Singer:
An Exploration of Holobiosis and Evolutionary Theory.” Biology and
Philosophy 32: 5–24.
Douglas, Angela E., and John H. Werren. 2016. “Holes in the Holo-
genome: Why Host-Microbe Symbioses Are Not Holobionts.” mBio 7 (2):
e02099–15.
Duvallet, Claire, Sean M. Gibbons, Thomas Gurry, Rafael A. Irizarry,
and Eric Alm. 2017. “Meta-Analysis of Gut Microbiome Studies
Identifies Disease-Specific and Shared Responses.” Nature Communications
8: 1784.
Eckburg, Paul B., Elisabeth M. Bik, Charles N. Bernstein, Elizabeth
Purdom, Les Dethlefsen, 等人. 2005. “Diversity of the Human Intestinal
Microbial Flora.” Science 308: 1635–1638.
Estrela, Sylvie, Benjamin Kerr, 和 J. Jeffrey Morris. 2016. “Transitions in
Individuality Through Symbiosis.” Current Opinion in Microbiology 31:
191–198.
Falony, Gwen, Sara Vieira-Silva, and Jeroen Raes. 2015. “Microbiology
Meets Big Data: The Case of Gut Microbiota-Derived Trimethylamine.”
Annual Review of Microbiology 6: 305–321.
Falony, Gwen, Marie Joossens, Sara Viera-Silva, Jun Wang, Youssef Darzi,
等人. 2016. “Population-Level Analysis of Gut Microbiome Variation.”
科学 352: 560–564.
Faust, Karoline, and Jeroen Raes. 2012. “Microbial Interactions: 从
Networks to Models.” Nature Reviews Microbiology 10: 538–550.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
259
Finucane, Mariel M., 托马斯·J. Sharpton, Timothy J. Laurent, 和
Katherine S. Pollard. 2014. “A Taxonomic Signature of Obesity in the
Microbiome? Getting to the Guts of the Matter.” PLOS One 9 (1): e84689.
Fischbach, Michael A., and Julia A. Segre. 2016. “Signaling in Host-
Associated Microbial Communities.” Cell 164: 1288–1300.
Fleissner, Christine K., Huebel, Nora, Mohamed M. A. El-Bary, Gunnar
Loh, Susanne Klaus, and Michael Blaut. 2010. “Absence of Intestinal
Microbiota Does Not Protect Mice From Diet-Induced Obesity.” British
Journal of Nutrition 104: 919–929.
Fuentes, Susana, Els van Nood, Sebastian Tims, Ineke H.-D. Jong, Cajo J. F.
ter Braak, 等人. 2014. “Reset of a Critically Disturbed Microbial
Ecosystem: Faecal Transplant in Recurrent Clostridium difficile Infection.”
ISME Journal 8: 1621–1633.
Gevers, Dirk, Subra Kugathasan, Lee A. Denson, Yoshiki Vázquez-Baeza,
Will Van Treuren, Boyu Ren, 等人. 2014. “The Treatment-Naïve
Microbiome in New-Onset Crohn’s Disease.” Cell Host & Microbe 15:
382–392.
吉尔伯特, Jack A., Robert A. 奎因, Justine Debelius, Zhenjiang Z. 徐,
James Morton, 等人. 2016. “Microbiome-Wide Association Studies
Link Dynamic Microbial Consortia to Disease.” Nature 535: 94–103.
Goodrich, Julia K., Sara C. Di Rienzi, Angela C. Poole, Omry Koren,
William A. Walters, 等人. 2014. “Conducting a Microbome Study.”
细胞 158: 250–262.
Gradmann, Christoph. 2000. “Isolation, Contamination, And Pure Culture:
Monomorphism and Polymorphism of Pathogenic Micro-Organisms as
Research Problem 1860–1880.” Perspectives on Science 9: 147–172.
绿色的, Sara, Maria Şerban, Raphael Scholl, Nicholaos Jones, Ingo Brigandt,
and William Bechtel. 2017. “Network Analyses in Systems Biology: 新的
Strategies for Dealing with Biological Complexity.” Synthese doi:10.1007/
s11229-016-1307-6.
Greenblum, Sharon, Hsuan-Chao Chiu, Roie Levy, Rogan Carr, and Elhanan
Borenstein. 2013. “Towards a Predictive Systems-Level Model of the
Human Microbiome: 进步, 挑战, and Opportunities.” Current
Opinion in Biotechnology 24: 810–820.
Greenblum, Sharon, 彼得·J. Turnbaugh, and Elhanan Borenstein. 2012.
“Metagenomic Systems Biology of the Human Gut Reveals Topological
Shifts Associated with Obesity and Inflammatory Bowel Disease.”
Proceedings of the National Academy of Sciences USA 109: 594–599.
Grouissin, Mathieu, Florent Mazel, Jon G. Sanders, Chris S. Smillie,
Sébastian Lavergne, 等人. 2017. “Unraveling the Processes Shaping
Mammalian Gut Microbiomes over Evolutionary Time.” Nature Commu-
nications 8: 14319.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
260
Methodological Strategies in Microbiome Research
Handelsman, Jo, Michelle R. Rondon, Sean F. 布雷迪, Jon Clardy, 和
Robert M. 古德曼. 1998. “Molecular Biological Access to the
Chemistry of Unknown Soil Microbes: A New Frontier for Natural
Products.” Chemistry & 生物学 5: R245–R249.
Handelsman, Jo. 2004. “Metagenomics: Application of Genomics to
Uncultured Microorganisms.” Microbiology and Molecular Biology Reviews
68: 669–685.
Harley, Isaac T.W., and Christopher L. Karp. 2012. “Obesity and the Gut
Microbiome.” Molecular Metabolism 1: 21–31.
Hooks, Katarzyna B., and Maureen A. O’Malley. 2017. “Dysbiosis and its
Discontents.” mBio 8: e01492–17.
Hooper, Lora V., and Jeffrey I. Gordon. 2001. “Commensal Host-Bacterial
Relationships in the Gut.” Science 292: 1115–1118.
Hooper, Lora V., Dan R. Littman, and Andrew J. Macpherson. 2012. “Inter-
actions Between the Microbiota and the Immune System.” Science 336:
1268–1273.
Howick, Jeremy. 2011. “Exposing the Vanities—and a Qualified Defense—
of Mechanistic Reasoning in Health Care Decision Making.” Philosophy of
科学 78: 926–940.
Hsiao, Elaine Y., Sara W. McBride, Sophia Hsien, Gil Sharon, Embriette
右. Hyde, 等人. 2013. “Microbiota Modulate Behavioral and Physio-
logical Abnormalities Associated with Neurodevelopmental Disorders.”
细胞 155: 1451–1463.
Huss, 约翰. 2014. “Methodology and Ontology in Microbiome Research.”
Biological Theory 9: 392–400.
Huttenhower, Curtis, Rob Knight, C. Titus Brown, J. Gregory Caporaso,
Jose C. Clemente, 等人. 2014. “Advancing the Microbiome Research
Community.” Cell 159: 227–230.
Jeffery, Ian B., Denise B. 林奇, and Paul W. O’Toole. 2016. “Composition
and Temporal Stability of the Gut Microbiota in Older Persons.” ISME
杂志 10: 170–182.
约翰逊, Elizabeth L., Stacey L. Heaver, William A. Walters, Ruth E. Ley.
2017. “Microbiome and Metabolic Disease: Revisiting the Bacterial
Phylum Bacteroidetes.” Journal of Molecular Medicine 95: 1–8.
Justus, James. 2008. “Complexity, Diversity, and Stability.” Pp. 321–350
in A Companion to the Philosophy of Biology. Edited by Sahotra Sahotra and
Anya Plutynski. 牛津: 布莱克威尔.
Kassam, Zain, Christine H. 李, Yuhong Yuan, and Richard H. 打猎. 2013.
“Fecal Microbiota Transplantation for Clostridium difficile: Systematic Review
and Meta-Analysis.” American Journal of Gastroenterology 108: 500–508.
Khoruts, 亚历山大, Johan Dicksved, Janet K. 扬松, and Michael J.
Sadowsky. 2010. “Changes in the Composition of the Human Fecal
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
261
Microbiome after Bacteriotherapy for Recurrent Clostridium difficile-
Associated Diarrhea.” Journal of Clinical Gastroenterology 44: 354–360.
Knights, Dan, Kara G. Lassen, and Ramnik J. Xavier. 2013. “Advances in
Inflammatory Bowel Disease Pathogenesis: Linking Host Genetics and
the Microbiome.” Gut 62: 1505–1510.
La Caze, 亚当. 2011. “The Role of Basic Science in Evidence-Based
Medicine.” Biology and Philosophy 26: 81–98.
Lawley, Trevor D., Simon Clare, Alan W. 沃克, 马克·D. Stares,
托马斯·R. Connor, 等人. 2012. “Targeted Restoration of the Intes-
tinal Microbiota with a Simple, Defined Bacteriotherapy Resolves
Relapsing Clostridium difficile Disease in Mice.” PLOS Pathogens 8 (10):
e1002995.
Lawley, Trevor D., and Alan W. 沃克. 2012. “Intestinal Colonization
Resistance.” Immunology 138: 1–11.
Layeghifard, Mehdi, 大卫·M. Hwang, 和大卫·S. Guttman. 2017.
“Disentangling Interactions in the Microbiome: A Network Perspective.”
Trends in Microbiology 25: 217–228.
Lepage, Patricia, Marion C. Leclerc, Marie Joossens, Stanislas Mondot,
Hervé M. Blottière, 等人. 2013. “A Metagenomic Insight into Our Gut’s
Microbiome.” Gut 62: 146–158.
征收, Maayan, Aleksandra A. Kolodziejczyk, Christoph A. Thaiss, 和
Eran Elinav. 2017. “Dysbiosis and the Immune System.” Nature Reviews
Immunolology 17: 219–232.
Lewis, James D., Eric Z. 陈, 罗伯特·N. Baldassano, Anthony R. Otley,
安妮·M. Griffiths, 等人. 2015. “Inflammation, Antibiotics, and Diet as
Environmental Stressors of the Gut Microbiome in Pediatric Crohn’s
Disease.” Cell Host & Microbe 18: 489–500.
Ley, Ruth E., 彼得·J. Turnbaugh, Samuel Klein, and Jeffrey I. Gordon.
2006. “Human Gut Microbes Associated with Obesity.” Nature 444:
1022–1023.
Louca, Stilianos, Laura W. Parfrey, and Michael Doebeli. 2016. “Decoupling
Function and Taxonomy in the Global Ocean Microbiome.” Science 353:
1272–1277.
Macpherson, Andrew J., Yasmin Köller, and Kathy D. McCoy. 2015. “这
Bilateral Responsiveness Between Intestinal Microbes and IgA.” Trends
in Immunology 36: 460–470.
庄园, Ohad, Roie Levy, and Elhanan Borenstein. 2014. “Mapping the
Inner Workings of the Microbiome: Genome- and Metagenomics-Based
Study of Metabolism and Metabolic Interactions in the Human Micro-
biome.” Cell Metabolism 20: 742–752.
Marchesi, Julian R. 2011. “Human Distal Gut Microbiome.” Environmental
Microbiology 13: 3088–3102.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
262
Methodological Strategies in Microbiome Research
Marino, Simeone, Nielson T. Baxter, Gary B. Huffnagle, Joseph F. Petrosino,
and Patrick D. Schloss. 2014. “Mathematical Modelling of Primary
Succession of Murine Intestinal Microbiota.” Proceedings of the National
Academy of Sciences U.S.A. 111: 439–444.
McCann, Kevin S. 2000. “The Diversity-Stability Debate.” Nature 405:
228–233.
Mendelsohn, J. 安德鲁. 2002. “‘Like All That Lives’: 生物学, 药品
and Bacteria in the Age of Pasteur and Koch.” History and Philosophy of
the Life Sciences 24: 3–36.
Momozawa, Yukihide, Valérie Deffontaine, Edouard Louis, and Juan F.
Medrano. 2011. “Characterization of Bacteria in Biopsies of Colon and
Stools by High Throughput Sequencing of the V2 Region of Bacterial
16S rRNA Gene in Human.” PLOS One 6 (2): e16952.
Moya, Andrés, and Manuel Ferrer. 2016. “Functional Redundancy-
Induced Stability of Gut Microbiota Subjected to Disturbance.” Trends
in Microbiology 24: 402–413.
Mullins, Thomas D., Theresa B. Britschgi, Robin L. Krest, and Stephen J.
Giovannoni. 1995. “Genetic Comparisons Reveal the Same Unknown
Bacterial Lineages in Atlantic and Pacific Bacterioplankton Communi-
ties.” Limnology and Oceanography 40: 148–158.
Noecker, Cecelia, Alexander Eng, Sujatha Srinivasan, Casey M. Thereiot,
Vincent B. Young, 等人. 2016. “Metabolic Model-Based Integration of
Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic
Links between Ecological and Metabolic Variation.” mSystems 1 (1):
e00013–15.
Odenbaugh, Jay. 2007. “Seeing the Forest and the Trees: Realism about
Communities and Ecosystems.” Philosophy of Science 74: 628–641.
Olesen, Scott W., and Eric J. Alm. 2016. “Dysbiosis Is Not an Answer.”
Nature Microbiology 1: 1–2.
Olsen, Gary J., David J. Lane, Stephen J. Giovannoni, and Norman R.
Pace. 1986. “Microbial Ecology and Evolution: A Ribosomal RNA
Approach.” Annual Review of Microbiology 40: 337–365.
O’Malley, Maureen A., Ingo Brigandt, Alan C. Love, 约翰·W. 克劳福德,
Jack A. 吉尔伯特, 等人. 2014. “Multilevel Research Strategies and Bio-
logical Systems.” Philosophy of Science 81: 811–828.
O’Malley, Maureen A. 2008. “Exploratory Experimentation and Scientific
实践: Metagenomics and the Proteorhodopsin Case.” History and
Philosophy of the Life Sciences 29: 337–358.
O’Malley, Maureen A. 2014. Philosophy of Microbiology. Cambridge UK:
剑桥大学出版社.
Parte, Aidan C. 2013. “LPSN—A List of Prokaryotic Names with Standing
in Nomenclature.” Nucleic Acids Research 42: D613–D616.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
263
彼得森, Charisse, and June L. 圆形的. 2014. “Defining Dysbiosis and
Its Influence on Host Immunity and Disease.” Cellular Microbiology 16:
1024–1033.
Prescott, Susan L. 2017. “History of Medicine: Origin of the Term Micro-
biome and Why It Matters.” Human Microbiome Journal 4: 24–25.
Rakoff-Nahoum, 赛斯, Kevin R. 促进, and Laurie E. Comstock. 2016.
“The Evolution of Cooperation Within the Gut Microbiota.” Nature
533: 255–259.
里德, Gregor, Jessica A. Younes, Henny C. Van der Mei, Gregory B. Gloor,
Rob Knight, and Henk J. Busscher. 2011. “Microbiota Restoration:
Natural and Supplemented Recovery of Human Microbial Communities.”
自然评论微生物学 9: 27–38.
Relman, David A., and Stanley Falkow. 2001. “The Meaning and Impact
of the Human Genome Sequence for Microbiology.” Trends in Micro-
生物学 9: 206–208.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
Ridaura, Vanessa K., Jeremiah J. Faith, Federico E. Rey, Jiye Cheng,
Alexis E. Duncan, 等人. 2013. “Gut Microbiota from Twins Dis-
cordant for Obesity Modulate Metabolism in Mice.” Science 341:
1241214.
Ross, Lauren N., and James F. 伍德沃德. 2016. “Koch’s Postulates: 一个
Interventionist Perspective.” Studies in History and Philosophy of Biological
and Biomedical Sciences 59: 35–46.
圆形的, June L., and Sarkis K. Mazmanian. 2009. “The Gut Microbiota
Shapes Intestinal Immune Responses During Health and Disease.” Nature
Reviews Immunology 9: 313–323.
Schulberg, J。, 和P. De Cruz. 2016. “Characterisation and Therapeutic
Manipulation of the Gut Microbiome in Inflammatory Bowel Disease.”
Internal Medicine Journal 46: 266–273.
Schwabe, Robert F., and Christian Jobin. 2013. “The Microbiome and
Cancer.” Nature Reviews Cancer 13: 800–812.
Schwiertz, Andreas, David Taras, Klaus Schäfer, Silvia Beijer, Nicolaas A.
Bos, 等人. 2010. “Microbiota and SCFA in Lean and Overweight
Healthy Subjects.” Obesity 18: 190–195.
Seedorf, Henning, Nicholas W. Griffin, Vanessa K. Ridaura, Alejandro
Reyes, Jiye Cheng, 等人. 2014. “Bacteria from Diverse Habitats Colonize
and Compete in the Mouse Gut.” Cell 159: 253266.
Singleton, Rivers Jr., and David R. Singleton. 2016. “Remembering Our
Forebears: Albert Jan Kluyver and the Unity of Life.” Journal of the History
of Biology 50: 169–218.
Skillings, Derek. 2016. “Holobionts and the Ecology of Organisms: 多-
Species Communities or Integrated Individuals?” Biology and Philosophy
31: 875–892.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
264
Methodological Strategies in Microbiome Research
Spor, Aymé, Omry Koren, and Ruth Ley. 2011. “Unravelling the Effects of
the Environment and Host Genotype on the Gut Microbiome.” Nature
Reviews Microbiology 9: 279–290.
Stahl, David A., David J. Lane, 加里·J. Olsen, and Norman R. Pace. 1985.
“Characterization of a Yellowstone Hot Spring Microbial Community by 5S
rRNA Sequences.” Applied and Environmental Microbiology 49: 1379–1384.
斯坦因, Jefferey L., Terence L. 沼泽, Ke Y. 吴, Hiroaki Shizuya, 和
Edward F. DeLong. 1996. “Characterization of Uncultivated Prokaryotes:
Isolation and Analysis of a 40-Kilobase-Pair Genome Fragment from a
Planktonic Marine Archaeon.” Journal of Bacteriology 178: 591–599.
Surana, Neeraj K., and Dennis L. Kasper. 2017. “Moving Beyond Microbiome-
Wide Associations to Causal Microbe Identification.” Nature 552: 244–247.
Sze, Marc A., and Patrick D. Schloss. 2016. “Looking for a Signal in the
Noise: Revisiting Obesity and the Microbiome.” mBio 7 (4): e01018–16.
唐, 瓦. H. Wilson, and Stanley L. Hazen. 2014. “The Contributory
Role of Gut Microbiota in Cardiovascular Disease.” Journal of Clinical
调查 124: 4204–4211.
Taxis, Tasia M., Sara Wolff, Sarah J. Gregg, Nicholas O. Minton, Chiqian
张, 等人. 2015. “The Players May Change but the Game Remains:
Network Analyses of Ruminal Microbiomes Suggest Taxonomic Differ-
ences Mask Functional Similarity.” Nucleic Acids Research 43: 9600–9612.
Turnbaugh, Peter J., Ruth E. Ley, Mahowald, 中号. A。, Magrini, 五、, Mardis,
乙. R。, and Jeffrey I. Gordon. 2006. “An Obesity-Associated Gut Microbiome
with Increased Capacity for Energy Harvest.” Nature 444: 1027–1031.
Turnbaugh, Peter J., Ruth E. Ley, Micah Hamady, Claire Fraser-Liggett,
Rob Knight, and Jeffrey I. Gordon. 2007. “The Human Microbiome
项目: Exploring the Microbial Part of Ourselves in a Changing World.”
自然 449: 804–810.
Turnbaugh, Peter J., Bäckhed, F。, Fulton, L。, and Jeffrey I. Gordon. 2008.
“Diet-Induced Obesity is Linked to Marked but Reversible Alterations
in the Mouse Distal Gut Microbiome.” Cell Host & Microbe 3: 213–223.
Turnbaugh, Peter J., Ridaura, V. K., Faith, J. J。, Rey, F. E., 骑士, R。,
and Jeffrey I. Gordon. 2009. “The Effect of Diet on the Human Gut
Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice.”
Science Translational Medicine 1 (6): 6ra14.
Tyson, Gene W., Jarrod Chapman, Philip Hugenholtz, Eric E. 艾伦,
Rachna J. Ram, 等人. 2004. “Community Structure and Metabolism
Through Reconstruction of Microbial Genomes from the Environment.”
自然 428: 37–43.
Van Nood, Els, Anne Vrieze, Max Nieuwdorp, Susana Fuentes, Erwin G.
Zoetendal, 等人. 2013. “Duodenal Infusion of Donor Feces for Recurrent
Clostridium difficile.” New England Journal of Medicine 368: 407–415.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3
科学观点
265
Venter, J. Craig, Karin Remington, 约翰·F. Heidelberg, Aaron L. Halpern,
Doug Rusch, 等人. 2004. “Environmental Genome Shotgun Sequencing
of the Sargasso Sea.” Science 304: 66–74.
Walters, William A., Zech Xu, and Rob Knight. 2014. “Meta-Analyses of
Human Gut Microbes Associated with Obesity and IBD.” FEBS Letters
588: 4223–4233.
王, Zeneng, Elizabeth Klipfell, 布莱恩·J. Bennett, Robert Koeth, 布鲁斯
S. Levison, 等人. 2011. “Gut Flora Metabolism of Phosphatidylcholine
Promotes Cardiovascular Disease.” Nature 472: 57–63.
Whitham, Thomas G., Joseph K. 贝利, Jennifer A. Schweitzer, 斯蒂芬·M.
Shuster, Randy K. Bangert, 等人. 2006. “A Framework for Community
and Ecosystem Genetics: From Gene to Ecosystems.” Nature Reviews
遗传学 7: 510–523.
Woese, Carl R., and George E. 狐狸. 1977. “Phylogenetic Structure of the
Prokaryotic Kingdom: The Primary Kingdoms.” Proceedings of the
National Academy of Sciences U.S.A. 74: 5088–5090.
吴, Gary D., and James D. Lewis. 2013. “Analysis of the Human Gut
Microbiome and Association with Disease.” Clinical Gastroenterology and
Hepatology 11: 774–777.
Yarza, Pablo, Pelin Yilmaz, Elmar Pruesse, Frank O. Glöckner, Wolfgang
路德维希, 等人. 2014. “Uniting the Classification of Cultured and
Uncultured Bacteria and Archaea Using 16S rRNA Gene Sequences.”
自然评论微生物学 12: 635–645.
张, Husen, John K. DiBaise, Andrea Zuccolo, Dave Kudrna, 米歇尔
Braidotti, 等人. 2009. “Human Gut Microbiota in Obesity and After
Gastric Bypass.” Proceedings of the National Academy of Sciences U.S.A.
106: 2365–2370.
赵, Liping. 2013. “The Gut Microbiota and Obesity: From Correlation
to Causality.” Nature Reviews Microbiology 11: 639–647.
朱, Weifei, Jill C. Gregory, Elin Org, Jennifer A. Buffa, Nilaksh Gupta,
等人. 2016. “Gut Microbial Metabolite TMAO Enhances Platelet Hyper-
reactivity and Thrombosis Risk.” Cell 165: 111–124.
Zoetendal, Erwin G., Chad T. 科利尔, Satoshi Koike, Roderick I. Mackie, 和
H. Rex Gaskins. 2004. “Molecular Ecological Analysis of the Gastro-
intestinal Microbiota: A Review.” Journal of Nutrition 134: 465–472.
我
D
哦
w
n
哦
A
d
e
d
F
r
哦
米
H
t
t
p
:
/
/
d
我
r
e
C
t
.
米
我
t
.
/
e
d
你
p
哦
s
C
/
A
r
t
我
C
e
–
p
d
我
F
/
/
/
/
2
6
2
2
3
9
1
7
9
0
3
8
5
p
哦
s
C
_
A
_
0
0
2
7
4
p
d
.
/
F
乙
y
G
你
e
s
t
t
哦
n
0
8
S
e
p
e
米
乙
e
r
2
0
2
3